Hyeshik Chang
The Transcriptomics Team employs high-throughput experimental methods to decode the intricate structure of multifaceted RNA regulatory mechanisms. The primary strategy centers around developing technologies that enable sensitive and unbiased investigation of RNA regulation at a single-molecule level throughout the cell. These methodologies are applied to uncover the molecular-level heterogeneity, shared features, patterns, and specificity of regulatory actions across diverse cellular compartments and processes. Active efforts are directed toward developing and refining techniques that facilitate detecting RNA-protein interactions, subcellular localization, and temporal labeling via single-molecule long-read sequencing. Machine learning and signal processing methodologies are utilized to identify these signals for further in-depth data analyses. The ultimate goal is to implement these new techniques across various cellular processes and biological systems, offering a comprehensive perspective on the molecular-level diversity of RNA regulatory actions within their unique physiological contexts.
Single-molecule RNA analysis at sub-nucleotide resolution
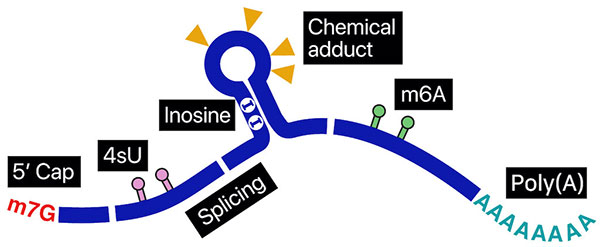
To fully comprehend the intricate regulatory mechanisms applied to RNAs, reading the rich information contained within individual full-length RNA molecules is essential. Nanopore direct sequencing provides opportunities to uncover previously elusive connections in genetic mechanisms. In nanopore direct RNA sequencing (DRS), a nucleic acid particle passes through a nanopore, a minuscule opening within a bilayer membrane. The passage of an RNA base through this opening uniquely disrupts the ionic current at different levels. This type of measurement provides a unique opportunity to investigate the minute details within an entire RNA molecule at a sub-nucleotide level. We are making tools to measure the length of poly(A) tails, find RNA modifications, report sites where proteins interact, and separate the sequenced molecules temporally in experimental timelines.
Full-length analysis of viral transcriptomes
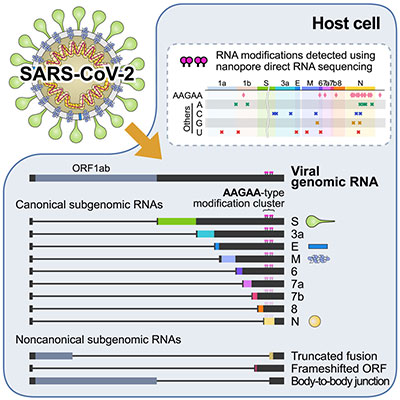
Investigating the evolving structure of transcriptomes in emerging viral strains is crucial. This is because new open reading frames and variants can transform the characteristics of viral proteins, such as their function and expression profiles. Moreover, viral RNAs can undergo chemical modifications such as methylation, deamination, and pseudouridylation. These modifications influence RNA stability, localization, and interactions with host proteins. Our research has involved detailed analysis of full-length transcriptomes and epitranscriptomes for several human coronaviruses, including SARS-CoV-2, human cytomegalovirus, and human hepatitis B virus. These analyses have shed light on the architecture of subgenomic transcripts, revealing potential RNA modifications. These modifications are indispensable to understanding how each virus shapes host-virus interactions.
 Center for RNA Research
Center for RNA Research
- IBS Center for RNA Research Room 531, Building 504, Seoul National University, 1, Gwanak-ro, Gwanak-gu, Seoul 08826 Tel. +82-2-887-2343 / Fax. +82-2-887-0244
Copyright(c) 2014 Center for RNA Research at IBS. All Rights Reserved.